The folly of a biological definition and staging in Parkinson’s disease
At the Pan-American Congress of the Movement Disorders Society in Cartagena, during the Hot Topics (Alberto J. Espay) and Controversies (Francisco Cardoso) plenary sessions, we sought to present the arguments of why a positive αSyn-SAA is an excellent test of aggregated α-synuclein pathology, aiding the clinical diagnosis of Parkinson’s disease but without informing Parkinson’s biology, clinical subtyping, biological subtyping, staging, or pathogenesis, nor monitor response to any treatments. Here is a summary of our arguments, integrated into five historical acts.
| |
Article Overview
Jump to:
|

Authors Francisco Cardoso and Alberto J. Espay during the 5th PAS Congress in Cartagena, Colombia in 2024.
Act 1. Pareidolia?
Heiko Braak and colleagues stained with α-synuclein immunostaining the brains of people with (n = 41) and without a history of Parkinson’s (n = 69) and ordered them from least to most staining. The static data was described as if in action, with Lewy pathology spreading from one brain region to another, and motor Parkinson’s making an entrance halfway through a six-stage cephalad “march” to the cortex.1 Although higher Braak stages were not aligned with aging, with clinical disability (Hoehn & Yahr), or with neuronal loss,2 they molded the clinical narrative so well those pesky details didn’t matter (Figure 1).
Corollary: The fable of active propagation of α-synuclein was born from creative pareidolia on the patterning of autopsy data points. The decade after the release of the Braak staging would be devoted to finding an underlying mechanism of the related Braak hypothesis.
Act 2. Infectivity?
Prions were conceived by Nobel Prize winner Stan Prusiner as proteins acting as ‘slow viruses,’ exhibiting infectious properties by replicating without nucleic acid. The idea of self-replicating proteins meant a biological information system parallel to the sequence-based central information transfer of molecular biology: from DNA to RNA to protein. Without a nucleic acid equivalent, the transfer of structural information must occur between proteins by templating themselves onto others. Prusiner suggested that the spread of brain pathology explaining neurodegeneration was via prions and introduced the term “α-synuclein prions” in 2015.3
Several questions were to be swiped under the carpet: If prion includes proteins’ active replication and propagation, shouldn’t the brain swell rather than shrink? Why are protein levels so low when measured in spinal fluid in ‘prionopathies’? Couldn’t the ‘prion behavior’ be alternatively explained by the physics of seeding, in which protein aggregation is a passive phase transformation under supersaturation conditions, proceeding along a thermodynamic gradient and independent of the amino acid sequence or capacity for templating?
These questions notwithstanding, the descriptor 'prion-like' became widely used in the vocabulary of Parkinson’s literature. It stuck as a new reality.
Corollary: Proteins morphing into virus-like agents offered an explanation to support the false but compelling Braak hypothesis. The next step was to create a test to reflect this notion.
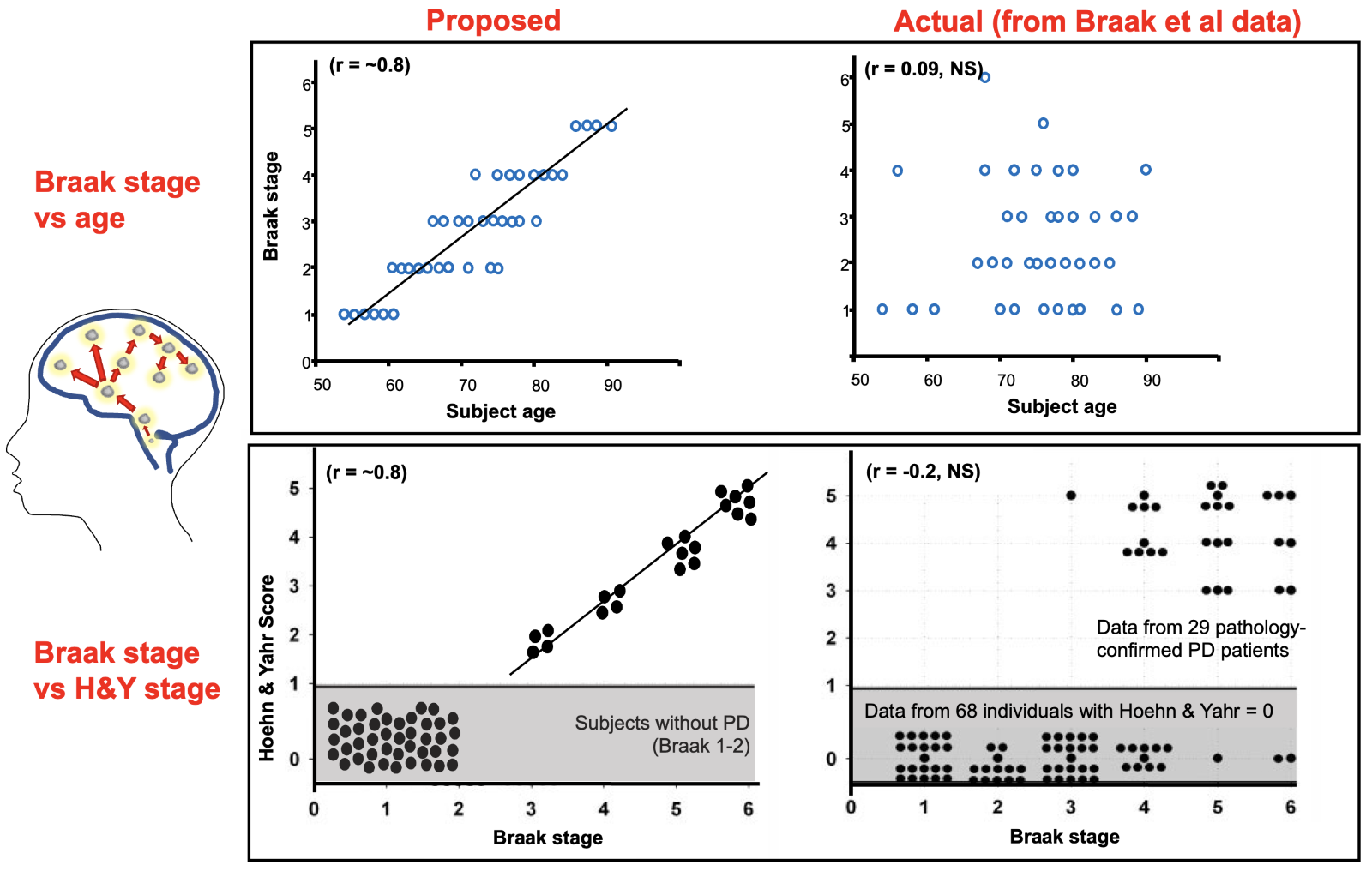 Figure 1. Braak Staging: Proposed relationships vs actual data. Braak and colleagues made static α-synuclein-staining data from unrelated brains appear dynamic and related. What is the problem with this linear “spread”? Pathology staging is not associated with aging or disability staging [Original data from Braak et al. Neurobiol Aging 2003; ;24:197–211, adapted by Burke, Dauer, and Vonsattel, Ann Neurol 2008;64:485-491]1, 2
Figure 1. Braak Staging: Proposed relationships vs actual data. Braak and colleagues made static α-synuclein-staining data from unrelated brains appear dynamic and related. What is the problem with this linear “spread”? Pathology staging is not associated with aging or disability staging [Original data from Braak et al. Neurobiol Aging 2003; ;24:197–211, adapted by Burke, Dauer, and Vonsattel, Ann Neurol 2008;64:485-491]1, 2
Act 3. Seeding that ‘amplifies’?
Once Parkinson’s disease was conceptualized as a prion-like disorder, the “self-replicating” mechanism attributed to the scrapie isoform of the prion protein, PrPSc, was also proposed to explain α-synuclein aggregation.4 Therefore, to detect α-synuclein in PD, it made sense to repurpose techniques designed to detect PrPSc in Creutzfeldt-Jakob disease, originally under the names real-time quaking-induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCA), and eventually seed amplification assay (SAA). If a “seed” templates many copies of itself, we should expect the products of the seed amplification to be identical to the seed.
This is not the case. The structures of the seeding products differ from the seeds from which they come.5 This is because seeding is a stochastic phenomenon governed by the same physical laws as crystallization; it depends on the microenvironment, not on any sequence of amino acids with ‘replicative’ properties.6 The seeding of PrP is very sensitive to PrP concentration, pH, and salt concentration. These microenvironmental conditions, particularly supersaturation, generically increase the intermolecular interactions, favoring the thermodynamic conditions for nucleation. Supersaturation means a concentration in the micromolar (mg/mL) range –1,000,000-fold greater than the human CSF in health and disease, which operates in picomolar (pg/ml) units. Once a specific supraphysiologic concentration threshold is reached, nucleation proceeds spontaneously and independently of the protein amino acid sequence or any other “templating code” within proteins. The result is stochastic protofilament stacking. That is, not strains, but infinitely varying polymorphs. For instance, two patients with the same ‘synucleinopathy’ (multiple system atrophy) have been shown to have different 2D folds in the ultrastructure of their aggregated alpha-synuclein (Figure 3B, 3C in Sawaya et al. 2021).7
Corollary: Amplification and strains are terms borrowed from virology after we conceived proteins as viruses in the making. But proteins never cease to behave like proteins: When a catalyst is present, they passively precipitate following the same physics of crystallization, without actual replication. The αSyn-SAA harnesses the physics of seeding to maximize the sensitivity of detecting seeds (aggregated protein). Despite its name, the assay does not amplify identical segments of protein or strains. Also, seeding in the laboratory does not mean seeding in the brain of the people tested – where supersaturation conditions do not exist (Figure 2).
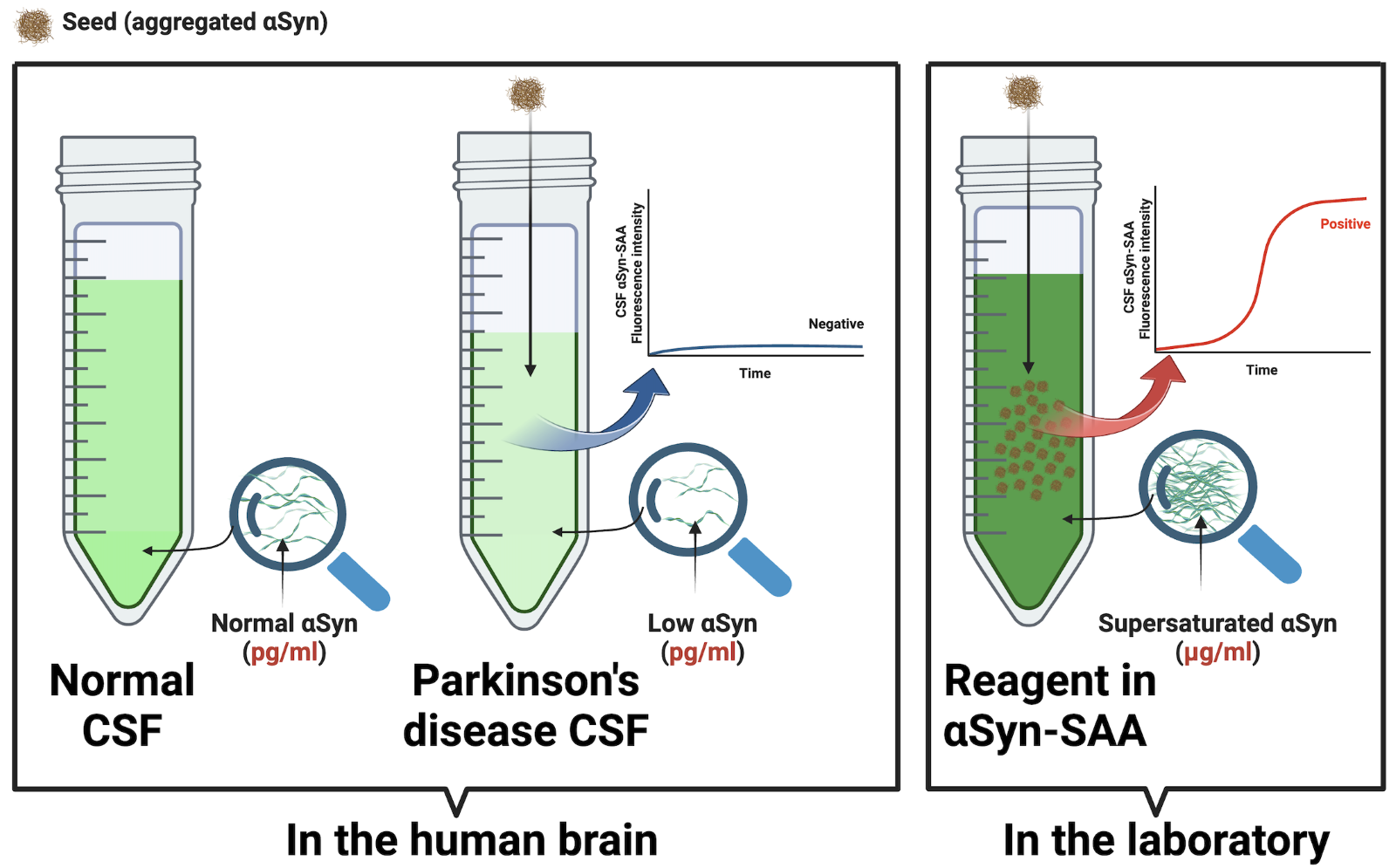 Figure 2. Supersaturation is requisite for the α-synuclein-seed amplification assay (αSyn-SAA). Seeding occurs in the laboratory only under supersaturation conditions (mg/ml) but not in the brain of people with Parkinson’s disease. The αSyn-SAA requires a concentration 1,000,000 times greater than in the human brain (pg/ml).
Figure 2. Supersaturation is requisite for the α-synuclein-seed amplification assay (αSyn-SAA). Seeding occurs in the laboratory only under supersaturation conditions (mg/ml) but not in the brain of people with Parkinson’s disease. The αSyn-SAA requires a concentration 1,000,000 times greater than in the human brain (pg/ml).
Act 4. Pathology as Biology?
A positive αSyn-SAA yields a sensitivity of 87.7% and a specificity of 96.3% for diagnosing Parkinson’s disease.8 This is an excellent performance for a diagnostic marker of aggregated α-synuclein, but inadequate to distinguish if the seeding catalyst comes from an individual with REM sleep behavior disorder, dementia with Lewy bodies, Parkinson’s disease, or a healthy control deploying their innate immune response system against a toxic or infectious exposure.9,10 As protein aggregation is a universal response to many insults, a positive αSyn-SAA cannot discriminate which biological, toxic, or infectious pathogen may have led to a positive test in the individual tested. Further, the αSyn-SAA cannot quantify the pathology burden nor the level of monomeric, soluble α-synuclein from which it comes. It is a qualitative test with only two possible outcomes: positive, meaning a seeding catalyst is present, or negative, meaning there is no catalyst. As such, the αSyn-SAA is extremely limited in its utility for understanding the pathophysiology of a particular patient or a group of patients. It cannot distinguish disease mechanisms or clinical phenotypes.
Case in point: in the Parkinson’s Progression Markers Initiative cohort, αSyn-SAA positivity did not correlate with motor severity or disease progression nor help distinguish autonomic dysfunction, cognitive impairment, depression, or any other clinical variable.8
Corollary: a positive αSyn-SAA sensitively supports, but does not replace, a clinical diagnosis of Parkinson’s disease, REM sleep behavior disorder, and dementia with Lewy bodies. It does not inform about biological subtypes, clinical severity, prognosis, or individualized pathogenesis. It cannot predict when, if ever, a normal individual might develop any clinical disorder. Lastly, a negative αSyn-SAA does not exclude Parkinson’s disease in a symptomatic individual.
Act 5. Positive αSyn-SAA as anchor of staging?
Act 1 revisited. Quantitative evaluation of pathology does not serve to stage Parkinson’s – but two newly proposed frameworks say a qualitative test of pathology does. A positive αSyn-SAA provides the “S+” for a biological classification of PD based on a triaxial ‘S-N-G’ scheme (synuclein, neurodegeneration, and genetic status),11 and a biological definition of ‘neuronal α-synuclein disease’ with a similarly triaxial ‘S-D-G’ (synuclein, dopamine dysfunction, and genetic status) ‘integrated stage system.’12 The ‘neuronal α-synuclein disease integrated stage system,’ in particular, is based on four non-validated assumptions:
- the presence of α-synuclein pathology alone equals ‘biologically defined’ disease;
- two positive biomarkers indicate worse disease staging than one;
- symptoms or signs cannot precede biomarker positivity;
- progression is unidirectional with stages based on the linear sequence of pathology identification, abnormal dopamine transporter SPECT scan, clinical symptoms, and ordinal categories of functional impairment (Figure 3).
The authors state, “It is probable that a substantial proportion of individuals in stage 1A — who are S+ but are asymptomatic — will not develop signs or symptoms and, therefore, might remain in stage 1A for life.”12 This admission compromises the logic of using such a staging system for early trial enrollment. Further, a mandatory requirement of any staging anchor is its correlation with the progression of the related condition. But neither genetic mutations nor αSyn-SAA positivity changes throughout the course of PD. This fact alone invalidates using them as staging markers. The immutability of genes and αSyn-SAA leaves only clinical anchors, without validated cut-offs for staging, from a clinical scale, the Movement Disorders Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS). Lastly, a positive αSyn-SAA cannot be used to reflect a patient’s response to treatment because any level of α-synuclein “seed” will make the test positive. The lack of change after cinpanemab, for instance, was recently confirmed.13
Corollary: A positive αSyn-SAA should not be relied upon to anchor a staging system or be considered to monitor response to treatments.
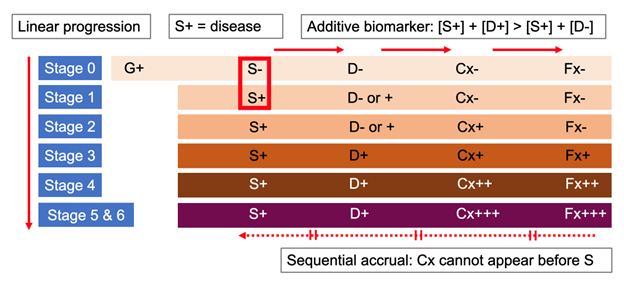
Figure 3. Key assumptions of the ‘integrated staging system’ for the biological definition of “neuronal α-synuclein disease” (Simuni et al, 2024).12 (1) S+ equals disease (S -, no disease or Stage 0); (2) two positive biomarkers indicate worse disease staging than one; (3) symptoms cannot precede biomarker positivity; and (4) progression is linear and sequential. G: genetic status; S: positive αSyn-seed amplification assay (αSyn-SAA); D: dopaminergic deficit by reduced SPECT imaging with ioflupane (I¹²³); Cx: clinical signs and symptoms; Fx: functional impairment; +, ++, +++: mild, moderate, severe.
Epilogue
Certain ideas have become so ingrained that they are deemed too integral to our field to be questioned, despite evidence to the contrary. Examples include MPTP as a model of Parkinson’s, SNCA mutations to explain a ‘virulent’ α-synuclein, the Braak staging system, and the prion-like hypothesis of pathology propagation.
Now, a troubling trend toward a 'biological definition' for Parkinson’s is poised to join this list. While a positive αSyn-SAA test can effectively indicate pathology presence and support (but not replace) clinical diagnoses, misconstruing it as indicative of pathogenesis, underlying biology, or disease staging is demonstrably untrue. No matter how compelling, ideas misrepresenting nature cannot form the foundation for effective treatments. The notion of 'α-synuclein prions' and the reverence given to αSyn-SAA testing are upheld by circular reasoning rooted in dogmas. It is imperative that we discard such concepts for the benefit of future patients. Synuclein isn't a virus or a toxin—it's a protein found throughout the body for a reason.14 Our focus should be restoring it to its normal state rather than destroying it once it becomes pathology.
References
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24(2):197-211. DOI: 10.1016/s0197-4580(02)00065-9.
- Burke RE, Dauer WT, Vonsattel JP. A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 2008;64(5):485-91. (In eng). DOI: 10.1002/ana.21541.
- Woerman AL, Stöhr J, Aoyagi A, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A 2015;112(35):E4949-58. (In eng). DOI: 10.1073/pnas.1513426112.
- Marotta NP, Ara J, Uemura N, et al. Alpha-synuclein from patient Lewy bodies exhibits distinct pathological activity that can be propagated in vitro. Acta neuropathologica communications 2021;9(1):188. (In eng). DOI: 10.1186/s40478-021-01288-2.
- Lövestam S, Schweighauser M, Matsubara T, et al. Seeded assembly in vitro does not replicate the structures of α-synuclein filaments from multiple system atrophy. FEBS Open Bio 2021;11(4):999-1013. (In eng). DOI: 10.1002/2211-5463.13110.
- Ezzat K, Sturchio A, Espay AJ. Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies. Biology (Basel) 2022;11(4) (In eng). DOI: 10.3390/biology11040535.
- Sawaya MR, Hughes MP, Rodriguez JA, Riek R, Eisenberg DS. The expanding amyloid family: Structure, stability, function, and pathogenesis. Cell 2021;184(19):4857-4873. (In eng). DOI: 10.1016/j.cell.2021.08.013.
- Siderowf A, Concha-Marambio L, Lafontant DE, et al. Assessment of heterogeneity among participants in the Parkinson's Progression Markers Initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol 2023;22(5):407-417. (In eng). DOI: 10.1016/s1474-4422(23)00109-6.
- Peelaerts W, Mercado G, George S, et al. Urinary tract infections trigger synucleinopathy via the innate immune response. Acta Neuropathol 2023;145(5):541-559. (In eng). DOI: 10.1007/s00401-023-02562-4.
- Allen Reish HE, Standaert DG. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 2015;5(1):1-19. (In eng). DOI: 10.3233/jpd-140491.
- Höglinger GU, Adler CH, Berg D, et al. A biological classification of Parkinson's disease: the SynNeurGe research diagnostic criteria. Lancet Neurol 2024;23(2):191-204. (In eng). DOI: 10.1016/s1474-4422(23)00404-0.
- Simuni T, Chahine LM, Poston K, et al. A biological definition of neuronal α-synuclein disease: towards an integrated staging system for research. Lancet Neurol 2024;23(2):178-190. (In eng). DOI: 10.1016/s1474-4422(23)00405-2.
- Hutchison RM, Fraser K, Yang M, et al. Cinpanemab in Early Parkinson Disease: Evaluation of Biomarker Results From the Phase 2 SPARK Clinical Trial. Neurology 2024;102(5):e209137. (In eng). DOI: 10.1212/wnl.0000000000209137.
- Espay AJ, Lees AJ. Loss of monomeric alpha-synuclein (synucleinopenia) and the origin of Parkinson's disease. Parkinsonism Relat Disord 2024:106077. (In eng). DOI: 10.1016/j.parkreldis.2024.106077.